The purpose of this study was to determine whether α-adrenergic receptor agonists have a role in alveolar fluid reabsorption, via Na,K-ATPase, in the alveolar epithelium. Alveolar fluid reabsorption increased approximately twofold with increasing concentrations of norepinephrine (NE) as compared with control rats. Treatment with the nonselective β-adrenergic receptor agonist, octopamine, and the specific α^sub 1^ agonist, phenylephrine, increased alveolar fluid reabsorption by 54 and 40%, respectively, as compared with control. The specific α^sub 1^-adrenergic receptor antagonist, prazosin, inhibited the stimulatory effects of NE by approximately 30%, whereas α^sub 2^-adrenergic antagonist, yohimbine, did not prevent the stimulatory effects of NE. The administration of ouabain, Na,K-ATPase inhibitor, prevented the NE-mediated increase in alveolar fluid reabsorption. In parallel with these changes, NE increased Na,K-ATPase activity and protein abundance in alveolar epithelial type II cells via the α^sub 1^- and β-adrenergic receptor. We report here that NE increased alveolar fluid reabsorption via the activation of both α^sub 1^- and β-adrenergic receptors, but not α^sub 2^-adrenergic receptors. These effects are due to increased activity and abundance of the Na,K-ATPase in the basolateral membrane of ATII cells.
Keywords: α-adrenergic receptors; active Na+ transport; alveolar fluid reabsorption; Na,K-ATPase; norepinephrine
Pulmonary edema develops as a result of changes in either the hydrostatic-oncotic pressure gradients across the pulmonary circulation or increased alveolo-capillary permeability (1). However, the resolution of alveolar fluid is effected by mechanisms that increase vectorial Na+ transport across the alveolo-capillary barrier, thus keeping the airspaces free of edema (2-6).
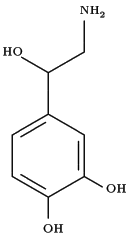
Norepinephrine (NE), a catecholamine with predominant α-adrenergic receptor effects, is commonly used in the treatment of patients with septic shock when fluid resuscitation fails to restore arterial blood pressure such as hypotensive states (7, 8). It has been reported that the infusion of NE to fetal sheep and neonatal guinea pigs decreased lung liquid production via the activation of a-adrenergic receptors (9, 10) and that β-adrenergic stimulation increases lung edema clearance by upregulation of Na+ channels and Na,K-ATPase (11-15). However, less is known about the effects of α-adrenergic receptor agonists on active Na+ transport. Several reports have shown that the administration of the α-adrenergic receptor agonists, NE and oxymetazoline, increased the activity of Na,K-ATPase in rat renal tubules (16, 17) and that during hypoxia endogenous norepinephrine increased net alveolar fluid clearance (18).
The purpose of this study was (1) to examine whether NE would increase active sodium transport and alveolar fluid reabsorption in adult rats; (2) to examine whether the effects on alveolar fluid reabsorption were mediated via the α-adrenergic receptors; (3) to investigate whether the effects of NE are inhibited by amiloride, which blocks apical Na+ channels, and ouabain, which inhibits the Na+ flux regulated by the basolaterally located Na,K-ATPase; and (4) to determine the effects of NE on the Na,K-ATPase in isolated alveolar type II cells.
METHODS
Isolated Perfused Lungs
The isolated perfused lung preparation used in our laboratory has been described in detail (6, 19, 20).
Study Croups
One hundred fifteen specific pathogen-free male Sprague-Dawley rats weighing 250-310 g were studied (Harlan Sprague-Dawley, Inc., Indianapolis, IN). All animals were provided food and water ad libitum and maintained on a 12 hour:12 hour light dark cycle. Norepinephrine, octopamine, phenylephrine, clonidine, prazosin, and yohimbine were purchased from Sigma Chemical Co. (St. Louis, MO). Ouabain and propranolol were purchased from ICN Biomedicals Inc. (Aurora, OH).
The experimental groups were as follows, with the number of animals in each group is given in parentheses:
Group A. Control group of isolated rat lungs studied at left atrial pressure (LAP) 0 cm H2O (n = 8).
Group B. NE (at varying concentrations) was perfused through the pulmonary circulation and the alveolar fluid reabsorption was determined: 10^sup -4^ M (n = 7), 10^sup -6^ M (n = 5), 10^sup -7^ M (n = 5), 10^sup -8^ M (n = 7), 10^sup -9^ M (n = 4), 10^sup -10^ M (n = 4).
Group C. To examine the contributory role of the apical Na+ channels and the basolateral Na,K-ATPase on NE effects, rat lungs were instilled with 10^sup -6^ M amiloride (Na+ channels blocker) either alone (n = 6) or in the presence of NE (n = 4). Also, rat lungs were perfused with 5 × 10^sup -4^ M ouabain (Na,K-ATPase blocker) either alone (n = 5) or in the presence of NE (n = 5).
Group D. To investigate which adrenergic receptor participates in the NE-mediated increase in alveolar fluid reabsorption, rat lungs were instilled and perfused with the selective α^sub 1^-adrenergic antagonist and selective α^sub 2^-adrenergic antagonist, 10^sup -5^ M prazosin (n = 4) and 10^sup -5^ M yohimbine (n = 4) respectively. Each substance was given alone and in the presence of 10^sup -6^ M NE (n = 4 and n = 5, respectively). To study the contribution of β-adrenergic receptor, rat lungs were treated with 10^sup -5^ M propranolol alone (n = 6) and in the presence of 10^sup -6^ M NE (n = 7). Rat lungs were also treated with prazosin and propranolol in the absence (n = 4) or presence of NE (n = 4).
Group E. The α-adrenergic receptors were selectively stimulated by instilling and perfusing rat lungs with 10^sup -6^ M octopamine, a nonselective α-adrenergic agonist (n = 5), 10^sup -6^ M phenylephrine, a selective α^sub 1^-adrenergic agonist (n = 6) and 10^sup -8^ M clonidine, an α^sub 2^-adrenergic agonist, (n = 4).
Isolation and Culture of Alveolar Type II Cells
ATII cells were isolated from pathogen-free male Sprague-Dawley rats (200-225 g), as previously described (20). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine scrum with 2 mM L-glutamine, 40 µg/ml gentamicin, 100 U/ml penicillin, and 100 µg/ml streptomycin and placed in culture for 2 days before the start of all experimental conditions.
Na,K-ATPase Activity
Ouabain-scnsitive ^sup 86^Rb^sup +^ uptake was used to estimate the rate of K+ transport by Na,K-ATPase in AEC (20). ^sup 86^Rb^sup +^ influx was quantified in aliquots of the SDS extract by liquid scintillation counting.
Preparation of Basolateral Plasma Membranes
Basolateral membranes (BLM) were purified as previously described (20).
Cell surface labeling. Briefly, cells were labeled EZ-link NHS-SS-biotin (Pierce Chemical Co., Rockford, IL). After labeling, cell lysates were incubated with streptavidin beads (Pierce Chemical Co.). The beads were thoroughly washed and then resuspended in Laemmli sample buffer solution as previously described (41). Proteins were analyzed by SDS-PAGE and Western blot.
Western blot analysis. Equal amounts of protein from BLMs were resolved by 10% SDS-PAGE and analyzed by immunoblotting with Na,K-ATPase anti-α1 (generous gift from M. Caplan, Yale University, New Haven, CT) monoclonal antibody.
Reverse Transcriptase-Polymerase Chain Reaction
The reverse transcriptase (RT) reaction was performed using the Superscript preamplication system by GIBCO-BRL (Gaithersburg, MD) following the manufacturer's instruction. One microgram of total RNA was converted into cDNA, after denaturing at 70°C for 15 min, by incubation with a buffer containing oligo-dT primers, the RT enzyme, and deoxynucleoside triphosphates (dNTPs) mix for 50 min at 42°C. The RT enzyme was then inactivated by incubation at 70°C for 15 min and the RNA removed by incubation with RNase H, for 20 min at 37°C. The resultant cDNAs were amplified by polymerase chain reaction (PCR) using α1D-and α2A-adrenergic receptor specific primers, then analyzed by 2% agarose gel electrophoresis.
Statistical Analysis
Data are presented as mean values ± SEM, n is the number of animals in each study group. One-way ANOVA was used when multiple comparisons were made followed by a multiple comparison test (Tukey) when the F statistic indicated significance. Results were considered significant when p
RESULTS
The lung of control rats instilled with 5 ml of buffered salt albumin solution cleared approximately 10% of the instillate in 1 hour (0.51 ± 0.02 ml/hour), whereas 10^sup -4^ to 10^sup -9^ M norepinephrine (NE) increased alveolar fluid reabsorption in a dose-dependent manner up to 100% above that in control lungs (Figure 1). Alveolar fluid clearance in rat lungs treated with 10^sup -10^ M NE was not different from control lungs. The rate of alveolar fluid reabsorption was similar regardless if the NE (^sup -6^ M) was added to the perfusate (0.92 ± 0.007 ml/hour) or to both the perfusate and instillate (0.87 ± 0.11 ml/hour). NE did not affect passive sodium or mannitol flux, or increase epithelial permeability to albumin (Table 1).
The NE-mediated increase in alveolar fluid reabsorption was regulated by both apical Na+ channels and basolateral Na, K-ATPases. As shown in Figure 2, instilling amiloride (10^sup -6^ M) to the airspaces decreased alveolar fluid reabsorption by approximately 30% in control rat lungs and inhibited the stimulatory effects of NE (10^sup -6^ M) by 55%. Also, perfusing ouabain (5 × 10^sup -4^ M) through the pulmonary circulation inhibited alveolar fluid reabsorption by approximately 60% in control rat lungs and by approximately 85% in NE (10^sup -10^ M)-treated rat lungs (Figure 2).
To investigate which α-adrenergic receptor regulated the NE-mediated increase in alveolar fluid reabsorption rat lungs were treated with the α^sub 1^-adrenergic receptor antagonist, prazosin, in the presence or absence of NE. Prazosin partially inhibited the NE-mediated increase in alveolar fluid clearance, but did not affect alveolar fluid reabsorption in control lungs (Figure 3). In contrast, the α^sub 2^-adrenergic receptor antagonist, yohimbine, did not inhibit the stimulatory effects of NE. The β-adrenergic receptor antagonist, propranolol, partially blocked the NE-mediated increase in alveolar fluid reabsorption (Figure 3). Finally, NE-mediated increased in alveolar fluid reabsorption was completely inhibited in rat lungs treated with both prazosin and propranolol.
To investigate the role of α-adrenergic receptor activation, rat lungs were instilled with the non-selective α-adrenergic receptor agonist, octopamine (10^sup -6^ M) and the specific α^sub 1^-adrenergic agonist, phenylephrine (10^sup -6^ M); both increased alveolar fluid reabsorption by approximately 60% and 55%, respectively. However, the specific α^sub 2^-adrenergic receptor agonist, clonidine 10^sup -8^ M, did not increase alveolar fluid reabsorption (Figure 4). None of the instilled agonists or antagonists had an effect on passive mannitol or sodium flux, or epithelial permeability for albumin (Table 1). However, the perfusate flow was reduced in rat lungs treated with NE and phenylephrine (Table 2). These effects are probably due to the pulmonary vasoconstriction induced probably by the activation of α^sub 1^-adrenergic receptors.
RT-PCR demonstrated the presence of messenger RNA for α-adrenergic receptors. An amplification product of the predicted size for the α-adrenergic receptor 1D (600 bp) and α-adrenergic receptor 2A (250 bp) was detected in the RT-PCR reactions using extracted ATII cell RNA (Figure 5A). Expression of the α-adrenergic receptor ID and α-adrenergic receptor 2C protein in ATII cell homogenates was analyzed by Western blot as shown in Figure 5B.
To investigate the role of α-adrenergic receptor activation on the Na,K-ATPase in alveolar epithelial type II (ATII) cells, cells were treated with NE (10^sup -6^ M), phenylephrine (10^sup -6^ M), α^sub 1^-adrenergic agonist, and clonidine (10^sup -8^ M), α^sub 2^-adrenergic receptor agonist, for 15 min at 37°C. As shown in Figure 6A, NE and phenylephrine increased the Na,K-ATPase activity by approximately 70% and 60%, respectively. However, the specific α^sub 2^-adrenergic receptor agonist, clonidine 10^sup -8^ M, did not increase Na,K-ATPase activity (Figure 6A). The NE-induced increase in Na,K-ATPase activity was regulated via α1- and β^sub 1^-adrenergic receptors, as pretreatment with either prazosin or propanolol partially inhibited the ouabain-inhibitable ^sup 86^Rb-uptake (Figure 6B). The phenylephrine-induced increase in Na,K-ATPase activity was inhibited by prazosin (aradrenergic receptor antagonist), but not by propanolol (β^sub 1^-adrenergic receptor angatonist).
The Na,K-ATPase α^sub 1^-subunit expression was significantly increased in the basolateral membrane of ATII cells treated with NE (Figure 7A). These results were confirmed by cell surface biotinylation experiments, which demonstrated that NE and phenylephrine both increased the number of Na,K-ATPase molecules in the plasma membrane. The NE-mediated increase in Na,K-ATPase protein abundance was mediated by both α^sub 1^- and β^sub 1^-adrenergic receptors, as pretreatment with prazosin and propranolol prevented the increase in Na,K-ATPase protein expression (Figure 7B). As expected, propanolol (β^sub 1^-adrenergic receptor antagonist) had no effect on the phenylephrine (α^sub 1^-adrenergic agonist) mediated increase in Na,K-pump abundance. The increased α^sub 1^-subunit abundance is not likely to represent increased de nova synthesis of Na,K-ATPase molecules because there was no change in Na,K-ATPase protein abundance in whole cell lysates (data not shown), which concords with a previous report (11, 40). Thus, we hypothesized that the increased number of Na,K-ATPase molecules within the basolateral membrane may be due to recruitment of existing Na,K-pumps from intracellular compartments.
DISCUSSION
Active Na+ transport and alveolar fluid reabsorption are important in maintaining the alveoli free of edema (4, 5, 15, 21). Several pharmacologic agents used in clinical practice have been found to regulate the vectorial Na+ transport, such as β-adrenergic receptor agonists (12, 18, 22, 23) and dopamine (3, 24). In the present study we demonstrate that NE increases active sodium transport and alveolar fluid reabsorption in an isolated perfused rat lung model, which is concordant with increases in Na,K-ATPase activity and protein abundance in isolated alveolar epithelial type II cells. These data are consistent with previous observations in neonatal lungs where NE decreased lung liquid production and induced reabsorption in guinea pigs (25) and sheep (26).
Norepinephrine is a nonselective catecholamine that has both α- and β-adrenergic activities (27, 28). Therefore, we performed three sets of studies in the isolated perfused lung to define the population of receptors involved in the NE-mediated increase in alveolar fluid reabsorption. First, we used NE and specific α^sub 1^- and α^sub 2^-adrenergic receptor antagonists (prazosin and yohimbine, respectively; Figure 3); second, we used specific α^sub 1^- and α^sub 2^-adrenergic receptor agonists (phenylephrine and clonidine, respectively; Figure 4); third, we used NE and propranolol to determine the role of β-adrenergic receptors (Figure 3). When coinstilled with NE, prazosin (the α^sub 1^-adrenergic antagonist) partially inhibited NE-stimulated alveolar fluid reabsorption, and phenylephrine (the α^sub 1^-adrenergic agonist) significantly increased alveolar fluid reabsorption. The aradrenergic antagonist, prazosin, had no effect on the baseline reabsorption of fluid. In contrast, the results do not support a role for the α^sub 2^-adrenergic receptor in the short-term regulation of sodium transport in the alveolar epithelium, as yohimbine (the α^sub 2^-adrenergic antagonist) did not inhibit the NE-stimulated alveolar fluid reabsorption, and clonidine (the α^sub 2^-adrenergic agonist) had no affect. As anticipated, propanolol partially inhibited the NE-stimulated alveolar fluid reabsorption. We have previously reported that β-adrenergic agonist, isoproterenol, stimulates active Na+ transport and alveolar fluid reabsorption in isolated perfused lungs (13, 20), and in cultured alveolar epithelial type 11 cells (11, 20). Isoproterenol is a nonselective β-adrenergic agonist with very low affinity for α-adrenergic receptors. We show here that NE stimulates alveolar fluid reabsorption to a similar extent as isoproterenol. Finally, the epithelial permeability to small solutes and to albumin was essentially unchanged by NE as compared with control rat lungs. The perfusate flow was slower in rats treated with NE and α-adrenergic agonists as compared with control rats, possibly due to pulmonary vasoconstriction caused by the stimulation of α^sub 1^- adrenergic receptors (29). Collectively, our data indicate that NE increases active Na+ transport and alveolar fluid reabsorption via the activation of both α^sub 1^- and β-adrenergic receptors, but not α^sub 2^-adrenergic receptors.
RT-PCR demonstrated the presence of both α^sub 1^- and α^sub 2^-adrenergic receptor mRNA in alveolar type II cells (Figure 5A). Western blot analysis confirmed that both α^sub 1^- and α^sub 2^-adrenergic receptor protein is expressed in alveolar type II cells. These data are consistent with other reports that used radioligand binding to detect the presence of β and α^sub 1^-adrenergic receptors in rat lung homogenates (30). Hasegawa and colleagues demonstrated that canine peripheral lungs express both α and β-adrenergic receptors, where 40% of the α-adrenergic receptors were α^sub 2^. Keeney and coworkers reported that α-adrenergic receptors are present in adult and neonatal rat alveolar epithelial type II cells (31). Also, it has been shown by mapping the mRNA distribution of α^sub 1^-adrenergic receptors in various rat tissues that the lung expresses α^sub 1A^ subtype (32). Our data is consistent with α^sub 1^-adrenergic receptor stimulation of active Na+ transport, but not with a previous study of Doe and colleagues, who reported that the mechanism by which NE decreased lung liquid production in fetal guinea pigs was by stimulating α^sub 2^-adrenergic receptors (25). It has been reported that fetal guinea pigs lungs at birth had a very rapid alveolar fluid clearance and that this increase was apparently mediated by endogenous catecholamines (34, 35). These discrepancies might stem from species and animal age differences (33). Of note, studies in the human lung will be needed to establish the relevance of the norepinephrine-mediated increase in lung function in the rodent model as it pertains to human lung pathophysiology.
To evaluate whether the stimulatory effects of NE were mediated via apical Na+ channels and the basolateral Na,K-ATPase, we treated rat lungs with amiloride and ouabain, respectively. As shown in Figure 2, the stimulatory effects of NE were inhibited by both amiloride and ouabain, suggesting that NE increased alveolar fluid reabsorption by upregulating the amiloride-sensitive Na+ channels and the ouabain sensitive Na,K-ATPase. These studies were then extended to examine the effects of NE on the Na,K-ATPase in ATII cells. We report here that NE stimulates Na,K-ATPase in alveolar epithelial cells via α^sub 1^-adrenergic receptor. As shown in Figure 6A, NE and phenylephrine, an α^sub 1^-adrenergic receptor agonist, increased the Na, K-ATPase activity in ATII cells, whereas ATII cells incubated with the α^sub 2^-adrenergic receptor agonist clondine showed no change in Na,K-pump activity. Supporting the notion that α^sub 1^-adrenergic receptors activation increases Na,K-ATPase activity is the report of Mallick and coworkers showing that NE increased Na,K-ATPase activity in rat brain via α^sub 1^-adrenergic receptor stimulation (36). In our study, NE-stimulated Na,K-ATPase activity by increasing the number of functioning Na,K-ATPase molecules in the basolateral membrane (Figures 6B). There are several mechanisms by which NE may increase the number of Na,K-ATPase molecules in the BLM, including changes in the rate of Na,K-ATPase protein synthesis. In fact, we have previously demonstrated that activation of the D2 receptor resulted in the increase in Na,K-ATPase abundance and enzymatic activity (37). However, this process occurred over a period of 18-24 h, much longer than the 15-minute time course of our experimental conditions. Therefore, we reasoned that Na,K-pumps which are stored in intracellular compartments were recruited for insertion in to the basolateral membrane. Although the signal transduction mechanisms were not examined in this report, several other reports have suggested that NE stimulation of α^sub 1^-adrenergic receptors activates phospholipase C via coupling to G^sub q^, proteins; phospholipase C, in turn catalyzes the hydrolysis of 1,2-diacylglycerol, which activates protein kinase C (PKC) (38). Stimulation of the β-adrenergic receptors activates the c-AMP protein kinase A (PKA) pathway (29). Both PKC and PKA pathways have been shown to regulate Na,K-ATPase activity (11, 39-41).
In conclusion, we report in this article that NE increased alveolar fluid reabsorption via the activation of both α^sub 1^- and β-adrenergic receptors, but not α^sub 2^-adrenergic receptors. These effects are due to upregulation of the Na,K-ATPase in the basolateral membrane of ATII cells and may be relevant for therapeutic strategies in patients with pulmonary edema.
References
1. Staub NC. Pulmonary edema. Physiol Rev 1974;54:678-811.
2. Azzam ZS, Sznajder JI. The cellular mechanisms contributing to lung edema clearance. Isr Med Assoc J 2000;2:235-239.
3. Barnard ML, Olivera WG, Rutschman DM, Bertorello AM, Katz AI, Sznajder JI. Dopamine stimulates sodium transport and liquid clearance in rat lung epithelium. Am J Respir Crit Care Med 1997;156:709-714.
4. Matalon S, O'Brodovich H. Sodium channels in alveolar epithelial cells: molecular characterization, biophysical properties, and physiological significance. Annu Rev Physiol 1999;61:627-661.
5. Malthay MA, Clerici C, Saumon G. Invited review: Active fluid clearance from the distal air spaces of the lung. J Appl Physiol 2002;93:1533-1541.
6. Rutschman DH, Olivera W, Sznajder JI. Active transport and passive liquid movement in isolated perfused rat lungs. J Appl Physiol 1993;75:1574-1580.
7. Martin C, Papazian L, Perrin G, Saux P, Gouin F. Norepinephrine or dopamine for the treatment of hyperdynamic septic shock? Chest 1993;103:1826-1831.
8. Schreuder WO, Schneider AJ, Groeneveld AB.Thijs LG. Effect of dopamine vs norepinephrine on hemodynamics in septic shock. Emphasis on right ventricular performance. Chest 1989;95:1282-1288.
9. Doe S, Woods B, Perks AM. Effects of norepinephrine on lung liquid production by in vitro lungs from fetal guinea pigs. Can J Physiol Pharmacol 1998;76:967-974.
10. Higuchi M, Murata Y, Miyake Y, Hesser J, Tyner J, Keegan KA, Porto M. Effects of norepinephrine on lung fluid flow rate in the chronically catheterized fetal lamb. Am J Obstet Gynecol 1987;157:986-990.
11. Bertorello AM, Ridge KM, Chibalin AV, Katz AI, Sznajder JI. Isoprotercnol increases Na+-K+-ATPase activity by membrane insertion of alpha-subunits in lung alveolar cells. Am J Physiol 1999;276:L20-L27.
12. Crandall ED, Heming TA, Palombo RL, Goodman BE. Effects of terbutaline on sodium transport in isolated perfused rat lung. J Appl Physiol 1986;60:289-294.
13. Saldías F, Lecuona E, Friedman E, Barnard ML, Ridge KM, Sznajder JI. Modulation of lung liquid clearance by isoproterenol in rat lungs. Am J Physiol 1998;274:L694-L701.
14. Saumon G, Basset G, Bouchonnet F, Crone C. cAMP and beta-adrenergic stimulation of rat alveolar epithelium. Effects on fluid absorption and paracellular permeability. Pflugers Arch 1987;410:464-470.
15. Sznajder JI. Strategies to increase alveolar epithelial fluid removal in the injured lung [editorial; comment]. Am J Respir Crit Care Med 1999;160:1441-1442.
16. Aperia A, Fryckstedt J, Holtbäck U, Belusa R, Cheng XJ, Eklöf AC, Li D, Wang ZM, Ohtomo Y. Cellular mechanisms for bi-directional regulation of tubular sodium reabsorption. Kidney Int 1996:49:1743-1747.
17. Ibarra F, Aperia A, Svensson LB, Eklöf AC, Grcengard P. Bidirectional regulation of Na+,K( + )-ATPase activity by dopamine and an alpha-adrenergic agonist. Proc Natl Acad Scu USA 1993;90:21-24.
18. Sakuma T, Hida M, Nambu Y, Osanai K, Toga H, Takahashi K, Ohya N, Inoue M, Wantanabe Y. Effects of hypoxia on alveolar fluid transport capacity in rat lungs. J Appl Physiol 2001;91:1766-1774.
19. Saldias F, Lecuona E, Friedman E, Barnard ML, Ridge KM, Sznajder JI. Modulation of lung liquid clearance by isoproterenol in rat lungs. Am J Physiol 1998;274:L694-L701.
20. Ridge KM, Olivera WG, Saldias F, Azzam Z, Horowitz S, Rutschman DH, Dumasius V, Factor P, Sznajder JI. Alveolar type 1 cells express the alpha2 Na,K-ATPase, which contributes to lung liquid clearance. Circ Res 2003;92:453-460.
21. Matthay MA, Folkesson HG, Verkman AS. Salt and water transport across alveolar and distal airway epithelia in the adult lung. Am J Physiol 1996;270:L487-L503.
22. Frank J, Wang Y, Osorio O, Matthay M. Beta-adrenergic agonist therapy accelerates the resolution of hydrostatic pulmonary edema in sheep and rats. J Appl Physiol 2000;89:1255-1265.
23. Berthiaume Y, Staub NC, Matthay MA. Beta-adrenergic agonists increase lung liquid clearance in anesthetized sheep. J Clin Invest 1987;79:335-343.
24. Saldias FJ, Lecuona E, Comelias AP, Ridge KM, Sznajder JI. Dopamine restores lung ability to clear edema in rats exposed to hyperoxia. Am J Respir Crit Care Med 1999;159:626-633.
25. Doc S, Perks AM. Alpha-adrenoreceptor influences on liquid movements by in vitro lungs from fetal guinea pigs. J Appl Physiol 1998;84:746-753.
26. Higuchi M, Murata Y, Miyake Y, Hesser J, Tyner J, Keegan KA, Porto M. Effects of norepinephrine on lung fluid flow rate in the chronically catheterized fetal lamb. Am J Obstet Gynecol 1987;157:986-990.
27. Ariens EJ, Simonis AM. Physiological and pharmacological aspects of adrenergic receptor classification. Biochem Pharmacol 1983;32:1539-1545.
28. Lefkowitz RJ, Caron MG, Stiles GL. Mechanisms of membrane-receptor regulation. Biochemical, physiological, and clinical insights derived from studies of the adrenergic receptors. N Engl J Med 1984;310:1570-1579.
29. Insel PA. Seminars in medicine of the Beth Israel Hospital, Boston. Adrenergic receptors: evolving concepts and clinical implications. N Engl J Med 1996:334:580-585.
30. Whitsett JA, Machulskis A, Noguchi A, Burdsall JA. Ontogeny of α 1- and β-adrenergic receptors in rat lung. Life Sci 1982;30:139-145.
31. Keeney SE, Oelberg DG. Alpha 1-adrenergic and muscarinic receptors in adult and neonatal rat type II pneumocytes. Lung 1993;171:355-366.
32. Faure C, Pimoule C, Arbilla S, Langer SZ, Graham D. Expression of alpha 1-adrenoceptor subtypes in rat tissues: implications for alpha 1-adrenoceptor classification. Eur J Pharmacol 1994;268:141-149.
33. Takayanagi I, Kawano K, Koike K. Alpha 2-adrenoceptor mechanisms in guinea-pig trachea. Eur J Pharmacol 1990;182:577-580.
34. Finley N, Norlin A, Baines DL, Folkesson HG. Alveolar epithelial fluid clearance is mediated by endogenous catecholamines at birth in guinea pigs. J Clin Invest 1998;101:972-981.
35. Norlin A, Finley N, Abedinpour P, Folkesson HG. Alveolar liquid clearance in the anesthetized ventilated guinea pig. Am J Physiol 1998;274:L235-L243.
36. Mallick BN, Adya HV, Faisal M. Norepinephrine-stimulated increase in Na+, K+-ATPase activity in the rat brain is mediated through alpha1A-adrenoceptor possibly by dephosphorylation of the enzyme. J Neurochem 2000;74:1574-1578.
37. Guerrero C, Ghosh A, Lecuona E, Ridge K, Santos E, Sznajder JI. Dopamine regulates NA,K-adenosine triphosphalase in alveolar epithelial cells via the milogen-activated protein kinase/extracellular-signal-regulated kinase pathway. Chest 1999;116:88S-89S.
38. Bylund DB, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz, RJ, Minneman KP, Molinoff PB, Ruffolo RR, Trendelenburg U. International Union of Pharmacology nomenclature of adrenoceptors. Pharmacol Rev 1994;46:121-136.
39. Bertorello AJ, Katz AI. Short-term regulation of renal Na,K-ATPase activity: physiological relevance and cellular mechanisms. Am J Physiol 1993;265:F743-F755.
40. Ridge, K. M., L. Dada, E. Lecuona, A. M. Bertorello, A. I. Katz, D. Mochly-Rosen, and J. I. Sznajder. 2002. Dopamine-induced exocytosis of Na,K-ATPase is dependent on activation of protein kinase C-epsilon and -delta. Mol Biol Cell 13:1381-1389.
41. Dada LA, Chandel NS, Ridge KM, Pedemonte C, Bertorello AM, Sznajder JI. Hypoxia-induced endocytosis of Na,K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J Clin Invest 2003;111:1057-1064.
Zaher S. Azzam, Yochai Adir, Astrid Crespo, Alejandro Comellas, Emilia Lecuona, Laura A. Dada, Norberto Krivoy, David H. Rutschman, Jacob I. Sznajder, and Karen M. Ridge
Medical Service, Veteran Affairs Chicago Health Care System; Division of Pulmonary and Critical Care Medicine, Northwestern University; and Northeastern Illinois University, Chicago, Illinois; Technion, Israel Institute of Technology, Haifa, Israel; and Universidad Central de Venezuela, Caracas, Venezuela
(Received in original form August 13, 2003; accepted in -final form July 9, 2004)
Supported in part by the Department of Veterans Affairs: VA-MREP, Parker B. Francis Foundation and NIH 48129.
Correspondence and requests for reprints should be addressed to Karen M. Ridge, Ph.D., Pulmonary and Critical Care Medicine, Northwestern University, Tarry 14-707, 300 E. Superior Street, Chicago, IL 60616. E-mail: kridge@northwestern.edu
This article has an online data supplement, which is accessible from this issue's table of contents online at www.atsjournals.org
Am J Respir Crit Care Med Vol 170. pp 730-736, 2004
Originally Published in Press as DOI: 10.1164/rccm.200308-1127OC on July 15, 2004
Internet address: www.atsjournals.org
Conflict of Interest Statement: Z.S.A. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; Y.A. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; A.C. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; A.C. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; E.L. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; L.A.D. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; N.K. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; D.H.R. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; J.I.S. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript; K.M.R. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
Copyright American Thoracic Society Oct 1, 2004
Provided by ProQuest Information and Learning Company. All rights Reserved




 Home
Home A
A Nabilone
Nabilone