Abstract
Homocysteine, a sulfur-containing amino acid, is a metabolite of the essential amino acid methionine, and exists at a critical biochemical intersection in the methionine cycle--between S-adenosylmethionine, the indispensable ubiquitous methyl donor, and vitamins B12 and folic acid. High blood levels of homocysteine signal a breakdown in this vital process, resulting in far-reaching biochemical and life consequences. The link between homocysteine and cardiovascular disease is well established, and decreasing plasma total homocysteine by providing nutritional cofactors for its metabolism has been shown to reduce the risk of cardiovascular events. Information has been emerging regarding a connection between homocysteine metabolism and cognitive function, from mild cognitive decline (age-related memory loss) to vascular dementia and Alzheimer's disease. Significant deficiencies in the homocysteine remethylation cofactors cobalamin (B12) and folate, as well as the trans-sulfuration cofactor vitamin B6, are commonly seen in the elderly population, with a resultant increase in homocysteine with advancing age. Hyperhomocysteinemia has been shown to be an independent risk factor for cognitive dysfunction. Indirect and direct vascular damage can be caused by homocysteine, which has been implicated in vascular dementia, with an increased risk of multiple brain infarcts and dementia as homocysteine levels rise. A significant correlation has been found between risk of Alzheimer's disease and high plasma levels of homocysteine, as well as low levels of folic acid, and vitamins B6 and B12. All of these disease associations are thought to be interrelated via increased homocysteine and S-adenosylhomocysteine and subsequent hypomethylation of numerous substances, including DNA and proteins, that render vascular structures and neurons more susceptible to damage and apoptosis. Providing the nutritional cofactors for proper functioning of the methionine cycle may improve methylation and protect the brain from damage. Further studies need to be performed to assess whether this will also reduce the risk of cognitive diseases and/or improve cognitive functioning.
Introduction
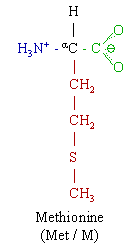
Homocysteine is a sulfur-containing amino acid produced in the metabolism of the essential amino acid methionine. It exists at a critical biochemical juncture between methionine metabolism and the biosynthesis of the amino acids cysteine and taurine. Homocysteine (Figure 1) is normally metabolized via two biochemical pathways--re-methylation, which converts homocysteine back to methionine, and trans-sulfuration, which converts homocysteine to cysteine and taurine. Abnormally high blood levels of homocysteine (hyperhomocysteinemia) signal a breakdown in this biochemical process, resulting in far-reaching biochemical and life consequences. For instance, hyperhomocysteinemia is a confirmed independent risk factor for cardiovascular disease, and has been epidemiologically and clinically implicated in a variety of other conditions, including neural tube defects, spontaneous abortion, placental abruption, low birth weight, osteoporosis, and neuropsychiatric disorders. It is thought that hyperhomocysteinemia might also contribute to the development of certain cancers. (1,2,3)
[FIGURE 1 OMITTED]
The correlative link between homocysteine and cardiovascular disease is well established. (4,5) High levels of homocysteine are correlated with significantly increased risk of coronary artery disease, (6-10) myocardial infarction, (11-15) cerebral occlusive disease (stroke), (16-18) and peripheral occlusive disease (deep vein thrombosis, intermittent claudication). (19-22) Lowering homocysteine by providing nutritional cofactors for its metabolism has been shown to reduce the risk of cardiovascular events. (23-25)
Less is known about the potential causal connection between hyperhomocysteinemia and brain dysfunction. This paper will explore the relationships between methionine, homocysteine, and their metabolites and cofactors, and cognitive decline, vascular dementia, and Alzheimer's disease (AD).
Methionine and Homocysteine Metabolism
Methionine, an amino acid utilized in protein formation, is the starting material for numerous other biochemical molecules. Methionine is converted via the enzyme methionine adenosyltransferase (MAT) to S-adenosylmethionine (SAMe), the most important methyl donor in the body. Methylation by SAMe is a critical step in the stabilization of many proteins, including myelin. Methylation of DNA stabilizes and protects this essential molecule, and can ultimately affect gene transcription coding for numerous proteins. SAMe is involved in the synthesis of other molecules, including creatine, methylcobalamin, phosphatidylcholine, melatonin, norepinephrine, coenzyme Q10, and carnitine. SAMe also participates in the formation of polyamines and in the metabolism of serotonin and niacinamide.
In hepatic phase II detoxification, SAMe contributes a methyl group for conjugation of a number of xenobiotics. In addition to this direct effect, proper methionine/SAMe/homocysteine metabolism is indirectly involved in other aspects of phase II detoxification. The downstream methionine metabolite taurine is necessary for amino acid and bile acid conjugation, and acylation reactions. Cysteine, another methionine metabolite, is necessary for sulfur conjugation and is a component of glutathione, a vital hepatic phase I and II component.
Regardless of the methyl-group acceptor involved, after donating its methyl group, SAMe becomes S-adenosylhomocysteine (SAH), which then becomes homocysteine after losing its adenosine (a reversible reaction catalyzed by the enzyme SAH hydrolase). Homocysteine must then either be: (1) recycled to methionine by taking on a methyl group provided by methylcobalamin, which is catalyzed by the enzyme methionine synthase; (2) recycled to methionine via the addition of a methyl group supplied by trimethylglycine (betaine), catalyzed by the zinc-dependent enzyme betaine-homocysteine methyltransferase; or (3) converted to the amino acids cysteine and taurine, which is catalyzed by the enzyme cystathione betasynthase, and with the help of the amino acid serine and pyridoxal 5'-phosphate (active vitamin B6) (Figure 2).
[FIGURE 2 OMITTED]
Optimal methionine metabolism is vital to a great number of biochemical processes in the body; however, dietary deficiencies, genetic enzyme polymorphisms, and acquired enzymatic blocks can negatively influence the normal metabolism of this essential amino acid (Table 1).
Dietary deficiencies of any of the cofactors involved in methionine and homocysteine metabolism (folic acid, vitamin B12, vitamin B6) can result in hyperhomocysteinemia. Folic acid is necessary in this process, as it acts as a methyl donor to cobalamin, forming methylcobalamin, which re-methylates homocysteine. Of the vitamin cofactors, folio acid deficiency seems to have the greatest effect on homocysteine levels, and folic acid supplementation (0.5-5 mg daily) results in a reduction of blood homocysteine of 25 percent, according to a 1998 meta-analysis. Addition of 0.5 mg vitamin B12 reduces homocysteine concentrations another seven percent. (23)
Overall, men have higher plasma homocysteine levels than women, although this difference becomes smaller with age and after menopause. (26-28) Other genetic influences include a polymorphism of the enzyme methylene tetrahydrofolate reductase (MTHFR), which catalyzes the last step in conversion of folic acid to its active form, 5-methyltetrahydrofolate (5MTHF). If an individual is homozygous for this genetic defect (C677T mutation of MTHFR), activity of this vital enzyme can decrease by 50 percent. Other genetic polymorphisms have been seen in hyperhomocysteinemia, including a defect in cystathione-beta synthase (CBS), which is involved in the vitamin B6-dependent trans-sulfuration pathway, and a defect in methionine synthase activity, which is involved in re-methylation of homocysteine.
Alterations in homocysteine metabolism can also be acquired, as is the case with certain dietary and lifestyle factors. Higher homocysteine levels have been found with increased coffee consumption, cigarette smoking, and chronic alcohol ingestion (interfering with methionine synthase activity). (29-31) In addition, nitrous oxide anesthesia irreversibly oxidizes the cobalt in vitamin B12, thereby rendering the vitamin inactive, which can severely affect methionine synthase-dependent homocysteine re-methylation. (32)
Homocysteine in Cognitive Decline
Memory loss is a common manifestation in the aging population, and it is a widely held belief that some amount of memory loss is inherent in the aging process. Whether memory loss is normal with age is debatable; however, extremes of cognitive decline, including dementia related to vascular disease (including post-stroke) and Alzheimer's disease, could never be termed "normal." The mechanisms of these cognitive deficits may be different, but it appears that metabolites and cofactors of methionine metabolism are involved.
Significant deficiencies in homocysteine re-methylation cofactors B12 and folate, as well as the trans-sulfuration cofactor vitamin B6, are commonly seen in the elderly population, (33-37) and have been found to contribute to a decline in cognitive function, as well as other neuropsychiatric disorders such as paresthesias, ataxia, sensory loss, and psychiatric disorders. (34,38))
Many methods and biochemical markers have been used to investigate these cofactors and their relevance to cognitive decline in the elderly, including serum B12, methylmalonic acid (a marker of B12 deficiency), serum folate, and plasma homocysteine. Total plasma homocysteine appears to be the most consistent marker of tissue deficiencies in nutrient cofactors, as well as cognitive performance in the elderly. (39)
Hyperhomocysteinemia was associated with poor recall in elderly subjects involved in the third National Health and Nutrition Examination Survey (NHANES III). (40) Participants, all over 60 years, with higher homocysteine levels scored lower in short-delayed recall tests. Lower folate levels were also associated with poor performance. Lower concentrations of B12 and folate, and higher levels of homocysteine, were directly associated with poor spatial copying skills in individuals ages 54-81 years in the Normative Aging Study. (41) Subjects with higher vitamin B6 levels scored better on two memory tests. Higher homocysteine levels were related to decreased performance in a battery of cognitive tests, including the Mini Mental State Examination (MMSE), in elderly individuals age 78 years who have been followed since 1932 when they were age 11. (42) This association was not seen in a cohort 15 years younger in the same study. In a sample of people age 55-75 years and of African-Caribbean descent, increased homocysteine concentrations were significantly correlated with cognitive impairment, independent of age, previous occupation, and risk factors of vascular disease (diabetes, hypertension, hyperlipidemia). (43)
Homocysteine levels were assessed in 32 healthy elderly individuals at baseline and five years later in an effort to determine if baseline homocysteine levels might predict cognitive decline. MMSE and one additional cognitive test (Alzheimer's disease Assessment Scale--ADAS-Cog) were performed at the start of the study and at the five-year follow-up. The mean age of participants was 79 at follow-up. Initial fasting total homocysteine accurately predicted worsening of MMSE and ADAS-Cog scores, signaling cognitive decline. (44)
Dementias have also been correlated with hyperhomocysteinemia. Nilsson et al found increased total plasma homocysteine in 45 percent of patients with dementia. Plasma homocysteine and blood folate levels directly correlated with severity of dementia in this study of 80 elderly patients, using a battery of dementia tests, including the Katz ADL index, the Berger scale, and a symptom score. (39)
Homocysteine and Vascular Dementia
After Alzheimer's disease, vascular dementia is the most common form of dementia, and accounts for 10-40 percent of dementia cases, depending on population, age, and diagnostic criteria. Vascular dementia is also known as multi-infarct dementia because the dementia is caused by multiple small brain infarcts. Hyperhomocysteinemia is an independent risk factor for stroke, (16,45,46) as well as for vascular dementia. (47-50)
In a study of 27 patients with hyperhomocysteinemia compared to 98 normal controls, Evers et al (51) found significantly increased blood pressure and microangiopathy, as well as a trend toward a higher rate of multiple infarcts, in the hyperholnocysteinemic group. In addition, the authors found impairment of cognitive processing, and believed the mechanism of these abnormalities involved microangiopathy; i.e., small vessel disease as compared to atherosclerotic changes in large vessels.
Of 336 consecutive patients seen at a memory clinic in Sweden, 20 percent were diagnosed with vascular dementia. This group exhibited the highest serum homocysteine levels, compared to Alzheimer's disease patients, mildly impaired subjects, and controls. The mildly impaired group (whom the authors termed the "dysmentia" group) also had significantly higher than normal levels, which may indicate homocysteine is a marker for a long-term process of cognitive worsening, possibly resulting in dementia. This study also noted that serum homocysteine was negatively associated with cognitive performance, assayed by the MMSE. (50)
The Rotterdam Study (52) did not find this correlation between homocysteine levels and dementia; however, it noted that patients with cognitive decline were older, less educated, and had a higher incidence of vascular disease and stroke. This prompted the authors to state that homocysteine could cause vascular damage leading to cognitive decline; however, their study only had a 2.7 year follow-up, which may have been too short a time period to detect a difference in the MMSE due to increasing homocysteine levels.
Alzheimer's Disease
A number of studies have recently found hyperhomocysteinemia is correlated with increased risk of Alzheimer's disease. In the Swedish study noted previously, (50) Lehmann et al found abnormally high homocysteine levels and poorer performance on the MMSE in AD patients versus non-demented controls. Hyperhomocysteinemia has been found to be associated with increased risk of AD in numerous other studies. (47,48,53-57)
In a recent paper by Nilsson et al, (53) plasma homocysteine was increased in late-onset AD, but not in early-onset AD. Homocysteine was also elevated in vascular dementia and was higher in late-onset AD patients with vascular disease, compared to other AD patients and controls. It was proposed that homocysteine is not causal to early-onset AD, and in late-onset AD is concomitant to other biochemical and vascular abnormalities that have a longer-term pathogenesis.
In another study, homocysteine levels were significantly higher in histologically confirmed AD patients, while folate and B12 levels were significantly lower than controls. The increase in homocysteine was small, revealing that a slight rise in this substance can have a profound impact on the risk of AD. Patients in the top third ([greater than or equal to] 14 [micro]mol/L) of homocysteine levels had a 4.5 times greater risk than those in the lower third ([less than or equal to] 11 [micro]mol/L). (54)
Researchers involved in the Framingham Study found a similar association between plasma homocysteine and AD. In a study published in the New England Journal of Medicine, Seshadri et al (56) assessed plasma homocysteine at baseline and eight years later in 1,092 elderly subjects and found higher levels of plasma homocysteine significantly more often in individuals who later developed AD than in those who did not develop AD. An increase in 5 [micro]mol/L increased the risk of AD by 40 percent. In persons with plasma homocysteine >14 [micro]mol/L, the risk doubled. This case-control study showed that plasma total homocysteine at baseline was an independent risk factor for AD.
An Australian study recently compared levels of homocysteine, B12, and folate in the plasma and cerebrospinal fluid (CSF) of AD patients compared to controls, and found significantly lower levels of B12 and folate in the plasma, along with increased levels of homocysteine in plasma and CSF in the AD patients. (58)
Is Homocysteine the Sole Culprit?
Homocysteine has been shown to be an independent risk factor for cognitive decline, vascular dementia, and AD. However, it is not certain homocysteine is the causal mechanism of these cognitive diseases. Since homocysteine is a metabolic byproduct of methionine metabolism, and since there are numerous nutrient cofactors involved in the methionine cycle, a deficiency of, or a metabolic dysfunction involving, any of these nutrients could affect how well homocysteine is either recycled or broken down. In addition, evidence points to these nutrients possibly independently affecting cognitive function.
In a study of serum folic acid and vitamin B12, Wang et al found low levels of folic acid, low levels of B12, and the combination all increased the relative risk of AD. A deficiency of either vitamin increased the risk 200 percent. (59) Low blood levels of folic acid and B12 were significantly associated with AD in 164 elderly AD patients. (54)
Nilsson et al found increased plasma homocysteine along with decreased serum folate, B12, and creatinine levels in 69 percent of demented and non-demented psychogeriatric patients. Serum folate was significantly lower in patients with dementia, compared with other patients. (60) In another study, 33 patients with mild-to-moderate dementia and hyperhomocysteinemia improved clinically after oral vitamin B12 (1 mg/ day) and folate (5 mg/day) for two months. MMSE scores were significantly improved in these patients, while severely demented patients did not improve. (61)
The Nun Study investigated blood levels of folate, B12, B6, cholesterol, and carotenoids in 95 elderly participants, age 77-98 years, 30 of whom died between baseline sampling and follow-up five years later. MMSE studies were also performed on patients at baseline. At autopsy, 50 percent of the nuns had a significant number of Alzheimer lesions in the brain, and a strong negative association was seen between serum folate and the severity of atrophy of the neocortex in those with AD. (62)
Neuropsychiatric symptoms related to vitamin B12 have been thought to be a result of long-standing, severe vitamin B12 deficiency accompanying anemia; however, researchers found neuropsychiatric disorders (paresthesias, sensory loss, dementia, ataxia, and psychiatric disorders) to be a common manifestation of deficiency without anemia or macrocytosis. These patients also had significant elevations in methylmalonic acid and homocysteine. Parenteral cobalamin supplementation resulted in significant improvements in these neuropsychiatric symptoms. (34) Treatment of cobalamin deficiency in dementia increased cerebral blood flow and improved symptomatology in 15 patients with mild to moderate dementia; however, severely demented patients did not improve. (63) A single-blind, placebo-controlled study showed improved cognition in elderly, cobalamin-deficient patients supplemented with B12 injections, (64) while two others did not. (65,66) Carmel et al noted B12 deficiency was a common finding in 16 patients with dementia. Parenteral cobalamin for four months improved concomitant neurological symptoms, but failed to improve dementia. (67)
Vitamin B12 deficiency and increasing severity of cognitive impairment has been seen in Alzheimer's disease patients compared to controls and patients with other dementias. (66,67) In a study of 52 AD patients, 50 hospitalized non-demented controls, and 49 elderly subjects living at home, AD patients were found to have the highest homocysteine levels and the highest methylmalonic acid levels, indicating a B12 deficiency. (57) In a Swedish study of 370 non-demented individuals followed for three years, the risk of development of AD was doubled in patients with low levels of the homocysteine-recycling cofactors B12 and folate. (59)
Miller et al were unsuccessful in attempting to find a statistical correlation between plasma homocysteine and AD; however, they did find low plasma pyridoxal 5'-phosphate (the active form of vitamin B6 in the blood and the vitamin cofactor involved in trans-sulfuration metabolism of homocysteine) was associated with increased incidence of AD. (70)
Significantly lower levels of SAMe have been found in cerebrospinal fluid (71) and the brain (72) in AD patients. In a small, open trial, high-dose oral SAMe (400 mg three times daily) improved mood and cognitive measures. (73)
Discussion
What are the mechanisms behind the connections among cognitive diseases and homocysteine, methionine, S-adenosylmethionine, and the nutritional cofactors involved in the methionine cycle'? Does homocysteine itself damage blood vessels and neurons, or is it a marker for deficiency or depletion of other interconnected molecules, or both?
There is evidence that hyperhomocysteinemia may promote dementia by more than one mechanism, including cerebral microangiopathy; (51) endothelial dysfunction; (72,73) oxidative stress; (74) neuronal DNA damage; (75,76) and enhancement of beta-amyloid peptide-mediated vascular smooth muscle toxicity, neurotoxicity, and apoptosis. (76-78) There is also a probability the homocysteine metabolite homocysteic acid--an N-methyl-D-aspartate agonist--can cause neuronal excitotoxicity and apoptosis. (70-81)
Even with the evidence presented above, the question still exists--does homocysteine have a causal connection to dementia, or is it a biomarker for other aberrant biochemical processes? The answer to this question may lie in one word--methylation. The immediate metabolite of methionine is SAMe, the most important methyl donor in the body. After SAMe donates its methyl group it becomes SAH, then homocysteine. If the biochemical cofactors of homocysteine metabolism are not present in sufficient quantities, or if there are other biochemical or genetic problems interfering with their production, transport, or utilization, the result is a rise in homocysteine levels. Increasing levels of homocysteine also result in a greater amount of SAH, as the reaction from SAH to homocysteine (catalyzed by SAH hydrolase) is a reversible reaction (Figure 3). SAH, as well as free adenosine from the metabolism of SAH, are potent inhibitors of transmethylation enzymes. SAH binds with high affinity to most SAMe-dependent methyltransferase enzymes, causing enzyme inhibition. (82) As stated above, this inhibition of methylation reactions can affect a great number of biochemical processes and can result in decreased production or metabolism of many substances, as well as decreased hepatocellular detoxification. For instance, high levels of homocysteine (and thus high levels of SAH) can decrease the activity of MTHFR, the enzyme responsible for methylating methylene tetrahydrofolate to 5MTHF, which then remethylates homocysteine via B12 using methionine synthase, another methyltransferase inhibited by SAH. (82)
[FIGURE 3 OMITTED]
SAH-induced inhibition of methyltransferase enzymes is very important in brain tissue because the alternate homocysteine re-methylating enzyme, betaine-homocysteine methyltransferase, does not have any activity in the brain (Figure 4). Also, only the liver, pancreas, kidney, and intestine exhibit full activity of the trans-sulfuration pathway that metabolizes homocysteine to cysteine and taurine.
[FIGURE 4 OMITTED]
It has been suggested, since CNS cells cannot export SAH from cells, that homocysteine extracellular transport is the only way CNS cells can decrease levels of SAH. (83) This can increase plasma homocysteine levels; however, homocysteine would not necessarily be the problem, but a marker for pathological levels of SAH. It is also possible SAH and homocysteine might be responsible for the pathogenesis of vascular diseases, neural tube defects, poor cognition, and other effects that previously have been attributed to homocysteine. It also may be that these abnormalities only affect individuals somehow genetically predisposed to their damage. Whatever the cause of abnormal levels of homocysteine and SAH may be, the results appear to be individualized and tissue specific.
Inhibited DNA methylation due to decreased folate has been shown to cause altered expression of certain genes, induction of cellular differentiation, alterations in chromatin conformation. and cell phenotypic changes. In neurodegenerative diseases such as mentioned above, homocysteine and SAH accumulation cause oxidative stress and neuronal excitotoxicity. Homocysteine also causes DNA strand breakage, resulting in mitochondrial membrane damage, nuclear disintegration, and apoptosis of neuronal cells. (81) Neuronal cells cultured in methionine- or folate-deficient growth medium exhibited significantly higher levels of apoptosis compared to controls. (76)
Another possible sequelae of hypomethylation involves amyloid-beta peptide, which accumulates in neurons of AD patients. The precursor of this peptide is amyloid precursor protein (APP), and the gene for this protein is highly methylated. Gene mutations involved in increased expression of APP and elevated production and extracellular deposition of the beta-amyloid peptide might be promoted by decreased methylation. (83,84) In addition, in cell cultures and in mice, folic acid deficiency and excess homocysteine impaired DNA repair in hippocampal neurons and sensitized them to amyloid-beta toxicity. (76)
Conclusion
It is vitally important that methyltransferase enzymes are fully active in the brain, as the brain is extremely dependent on the methyl groups provided by folic acid, vitamin B12, and SAMe. However, these methyl group providers can be stressed in many situations--dietary deficiency, poor absorption, improper conversion of folates, defective transport and/or oxidative conversion of folate and/or B12, and genetic polymorphisms of the MTHFR enzyme. "Pushing" these enzymes by providing large amounts of their cofactors appears to be an effective way of combating the reduction of enzyme activity often seen in these individuals.
Studies of individuals with a wide range of cognitive impairment consistently show increased plasma homocysteine and decreases in enzymatic cofactors involved in methionine and homocysteine metabolism. The few studies available that have supplemented demented patients with these cofactors have usually given only one cofactor for short periods of time, with mixed results. Given the apparent individualized metabolic dysfunctions in dementia, it makes sense to provide all the nutrients needed for the metabolism of these amino acids.
In the small number of intervention studies seen in the scientific literature, patients who respond best appear to be those with mild or moderate dementia. It remains to be seen if higher doses or longer-term studies will result in better efficacy in more severely affected patients. Deficiency of these vitamin cofactors is a common occurrence in the elderly. Since B12, folate, betaine, and B6 have been proven to lower homocysteine levels and are an inexpensive intervention, supplementation is a prudent preventive measure in middle-aged to elderly individuals. Preventively, 1 mg B12, 800 mcg folate, 25-50 mg B6, and 500-1,000 mg betaine can be utilized in at-risk individuals, while a regimen of 2 mg B12, 25 mg folate, 50-100 mg B6, and 1-3 g betaine appears to be a rational approach for treatment of those with dementia.
References
(1.) Miller AL, Kelly GS. Homocysteine metabolism: nutritional modulation and impact on health and disease. Altern Med Rev 1997;2:234-254.
(2.) Urnov FD. Methylation and the genome: the power of a small amendment. J Nutr 2002;132:2450S-2456S.
(3.) Zhu BT. On the mechanism of homocysteine pathophysiology and pathogenesis: a unifying hypothesis. Histol Histopathol 2002;17:1283-1291.
(4.) Bautista LE, Arenas IA, Penuela A, Martinez LX. Total plasma homocysteine level and risk of cardiovascular disease: a meta-analysis of prospective cohort studies. J Clin Epidemiol 2002;55:882-887.
(5.) No authors listed. Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA 2002;288:2015-2022.
(6.) Hopkins P, Wu L, Wu J, et al. Higher plasma homocyst(e)ine and increased susceptibility to adverse effects of low folate in early familial coronary artery disease. Arterioscler Thromb Vasc Biol 1995;15:1314-1320.
(7.) Loehrer F, Angst C, Haefeli W, et al. Low whole-blood S-adenosylmethionine and correlation between 5-methyltetrahydrofolate and homocysteine in coronary artery disease. Arterioscler Thromb Vasc Biol 1996;16:727-733.
(8.) Boushey C, Beresford S, Omenn G, Motulsky A. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA 1995;274:1049-1057.
(9.) Robinson K, Mayer E, Miller D, et al. Hyperhomocysteinemia and low pyridoxal phosphate. Common and independent reversible risk factors for coronary artery disease. Circulation 1995;92:2825-2830.
(10.) Nikfardjam M, Graf S, Hornykewycz S, et al. Homocysteine plasma levels in young patients with coronary artery disease. Relation to history of acute myocardial infarction and anatomical extent of disease. Thromb Res 2001;103:S35-S39.
(11.) Landgren F. Israelsson B, Lindgren A, et al. Plasma homocysteine in acute myocardial infarction: homocysteine-lowering effect of folic acid. J Int Med 1995;237:381-388.
(12.) Chasan-Taber L, Selhub J, Rosenberg I, et al. A prospective study of folate and vitamin B6 and risk of myocardial infarction in US physicians. J Am Coll Nutr 1996;15:136-143.
(13.) Knekt P, Alfthan G, Aromaa A, et al. Homocysteine and major coronary events: a prospective population study amongst women. J Intern Med 2001;249:461-465.
(14.) Whincup PH, Refsum H, Perry IJ, et al. Serum total homocysteine and coronary heart disease: prospective study in middle aged men. Heart 1999;82:448-454.
(15.) Bots ML, Launer LJ, Lindemans J, et al. Homocysteine and short-term risk of myocardial infarction and stroke in the elderly: the Rotterdam Study. Arch Intern Med 1999;159:38-44.
(16.) Brattstrom L, Lindgren A, Israelsson B, et al. Hyperhomoeysteinaemia in stroke: prevalence, cause, and relationships to type of stroke and stroke risk factors. Eur J Clin Invest 1992;22:214-221.
(17.) Perry IJ, Refsum H, Morris RW, et al. Prospective study of serum total homocysteine concentration and risk of stroke in middle-aged British men. Lancet 1995;346:1395-1398.
(18.) Hultberg B, Andersson A, Lindgren A. Marginal folate deficiency as a possible cause of hyperhomocystinaemia in stroke patients. Eur J Clin Chem Clin Biochem 1997;35:25-28.
(19.) Molgaard J, Malinow MR, Lassvik C, et al. Hyperhomocyst(e)inaemia: an independent risk factor for intermittent claudication. J Intern Med 1992;231:273-279.
(20.) Cheng SW, Ting AC, Wong J. Fasting total plasma homocysteine and atherosclerotic peripheral vascular disease. Ann Vase Surg 1997;11:217-223.
(21.) den Heijer M, Koster T, Blom HJ, et al. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N Engl J Med 1996;334:759-762
(22.) Beaumont V, Malinow MR, Sexton G, et al. Hyperhomocyst(e)inemia, anti-estrogen antibodies and other risk factors for thrombosis in women on oral contraceptives. Atherosclerosis 1992;94:147-152.
(23.) No authors listed. Lowering blood homocysteine with folic acid based supplements: meta-analysis of randomised trials. Homocysteine Lowering Trialists' Collaboration. BMJ 1998;316:894-898.
(24.) Schnyder G, Roffi M, Flammer Y, et al. Effect of homocysteine-lowering therapy with folic acid, vitamin B12, and vitamin B6 on clinical outcome after percutaneous coronary intervention: the Swiss Heart study: a randomized controlled trial. JAMA 2002;288:973-979.
(25.) Yap S, Boers GH, Wilcken B, et al. Vascular outcome in patients with homocysteinuria due to cystathione beta-synthase deficiency treated chronically: a multicenter observational study. Arterioscler Thromb Vasc Biol 2001;21:2080-2085.
(26.) Wouters MGAJ, Moorrees MTEC, Van der Mooren MJ, et al. Plasma homocysteine and menopausal status. Eur J Clin Inwest 1995;25:801-805.
(27.) Brattstrom L, Lindgren A, Isrealsson B, et al. Homocysteine and cysteine: determinants of plasma levels in middle-aged and elderly subjects. J Intern Med 1994;236:633-641.
(28.) Nygard O, Vollset SE, Refsum H, et al. Total plasma homocysteine and cardiovascular risk profile--the Hordaland homocysteine study. JAMA 1995;274:1526-1533.
(29.) Nygard O, Refsum H, Ueland PM, et al. Coffee consumption and plasma total homocysteine: The Hordaland Homocysteine Study. Am J Clin Nutr 1997;65:136-143.
(30.) Cravo ML, Gloria LM, Selhub J, et al. Hyperhomocysteinemia in chronic alcoholism: correlation with folate, vitamin B-12, and vitamin B-6 status. Am J Clin Nutr 1996;63:220-224.
(31.) Hultberg B, Berglund M, Andersson A, Frank A. Elevated plasma homocysteine in alcoholics. Alcohol Clin Exp Res 1993:17:687-689.
(32.) Flippo TS, Holder WD Jr. Neurologic degeneration associated with nitrous oxide anesthesia in patients with vitamin B12 deficiency. Arch Surg 1993:128:1391-1395.
(33.) Quinn K, Basu TK. Folate and vitamin B12 status of the elderly. Eur J Clin Nutr 1996;50:340-342.
(34.) Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med 1988;318:1720-1728.
(35.) Lindenbaum J, Rosenberg IH, Wilson PW, et al. Prevalence of cobalamin deficiency in the Framingham elderly population. Am J Clin Nutr 1994;60:2-11.
(36.) Joosten E, van den Berg A, Riezler R, et al. Metabolic evidence that deficiencies of vitamin B-12 (cobalamin), folate, and vitamin B-6 occur commonly in elderly people. Am J Clin Nutr 1993;58:468 476.
(37.) Pennypacker LC, Allen RH, Kelly JR et al. High prevalence of cobalamin deficiency in elderly outpatients. J Am Geriatr Soc 1992;40:1197-1204.
(38.) Metz J, Bell AH, Flicker L, et al. The significance of subnormal serum vitamin B12 concentration in older people: a case control study. J Am Geriatr Soc 1996;44:1355-1361.
(39.) Nilsson K, Gustafson L, Hultberg B. The plasma homocysteine concentration is better than that of serum methylmalonic acid as a marker for sociopsychological performance in a psychogeriatric population. Clin Chem 2000;46:691-696.
(40.) Morris MS, Jacques PF, Rosenberg IH, Selhub J. Hyperhomocysteinemia associated with poor recall in the third National Health and Nutrition Examination Survey. Am J Clin Nutr 2001;73:927-933.
(41.) Riggs KM, Spiro A 3rd, Tucker K, Rush D. Relations of vitamin B-12, vitamin B-6, folate, and homocysteine to cognitive performance in the Normative Aging Study. Am J Clin Nutr 1996;63:306-314.
(42.) Duthie SJ, Whalley LJ, Collins AR, et al. Homocysteine, B vitamin status, and cognitive function in the elderly. Am J Clin Nutr 2002;75:908-913.
(43.) Stewart R, Asonganyi B, Sherwood R. Plasma homocysteine and cognitive impairment in an older British African-Caribbean population. J Am Geriatr Soc 2002:50:1227-1232.
(44.) McCaddon A, Hudson R Davies G, et al. Homocysteine and cognitive decline in healthy elderly. Dement Geriatr Cogn Disord 2001;12:309-313.
(45.) Boers GH, Smals AG, Trijbels FJ, et al. Heterozygosity for homocysteinuria in premature peripheral and cerebral occlusive arterial disease. N Engl J Med 1985;313:709-715.
(46.) Pasini FL, Frigerio C, Petri S. diPerri T. Plasma homocysteine in ischemic stroke. Stroke 1995;6:795-800.
(47.) McIlroy SP, Dynan KB, Lawson JT, et al. Moderately elevated plasma homocysteine, methylenetetrahydrofolate reductase genotype, and risk for stroke, vascular dementia, and Alzheimer disease in Northern Ireland. Stroke 2002;33:2351-2356.
(48.) Leblhuber F, Walli J. Homocysteine and B vitamins in dementia. Am J Clin Nutr 2001;73:127-134. [letter]
(49.) Bertsch T, Mielke O, Holy S, et al. Homocysteine in cerebrovascular disease: an independent risk factor for subcortical vascular enechalopathy. Clin Chem Lab Med 2001;39:721-724.
(50.) Lehmann M, Gottfries CG, Regland B. Identification of cognitive impairment in the elderly: homocysteine is an early marker. Dement Geriatr Cogn Disor 1999;10:12-20.
(51.) Evers S, Koch H, Grotemeyer K, et al. Features, symptoms, and neurophysiological findings in stroke associated with hyperhomocysteinemia. Arch Neurol 1997;54:1276-1282.
(52.) Kalmijn S, Launer LL, Lindemans J, et al. Total homocysteine and cognitive decline in a community-based sample of elderly subjects. Am J Epidemiol 1999;150:283-289.
(53.) Nilsson K, Gustafson L, Hultberg B. Relationship between plasma homocysteine and Alzheimer's disease. Dement Geriatr Cogn Disord 2002; 14:7-12.
(54.) Clarke R, Smith AD, Jobst KA, et al. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch Neurol 1998;55:1449-1455.
(55.) McCaddon A, Davies G, Hudson P, et al. Total serum homocysteine in senile dementia of Alzheimer type. Int J Geriatr Psychiatry. 1998;13:235-239.
(56.) Seshadri S, Beiser A, Selhub J, et al. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N Engl J Med 2002;346:476-483.
(57.) Joosten E, Lesaffre E, Riezler R, et al. Is metabolic evidence for vitamin B-12 and folate deficiency more frequent in elderly patients with Alzheimer's disease? J Gerontol A Biol Sci Med Sci 1997;52:M76-M79.
(58.) Selley ML, Close DR, Stern SE. The effect of increased concentrations of homocysteine on the concentration of (E)-4-hydroxy-2-nonenal in the plasma and cerebrospinal fluid of patients with Alzheimer's disease. Neurobiol Aging 2002;23:383-388.
(59.) Wang HX, Wahlin A, Basun H, et al. Vitamin B(12) and folate in relation to the development of Alzheimer's disease. Neurology 2001;56:1188-1194.
(60.) Nilsson K, Gustafson L, Faldt R, et al. Hyperhomocysteinemia--a common finding in a psychogeriatric population. Eur J Clin Invest 1996;26:853-859.
(61.) Nilsson K, Gustafson L, Hultberg B. Improvement of cognitive functions after cobalamin/ folate supplementation in elderly patients with dementia and elevated plasma homocysteine. bit J Geriatr Psychiatry 2001;16:609-614.
(62.) Snowdon DA, Tully CL, Smith CD, et al. Serum folate and the severity of atrophy of the neocortex in Alzheimer disease: findings from the Nun Study. Am J Clin Nutr 2000;71:993-998.
(63.) Nilsson K, Warkentin S, Hultberg B, et al. Treatment of cobalamin deficiency in dementia, evaluated clinically and with cerebral blood flow measurements. Aging (Milano) 2000;12:199-207.
(64.) Van Asselt DZ, Pasman JW, van Lier HJ, et al. Cobalamin supplementation improves cognitive and cerebral function in older, cobalamin-deficient persons. J Gerontol A Biol Sci Med Sci 2001;56:M775-M779.
(65.) Kwok T, Tang C, Woo J, et al. Randomized trial of the effect of supplementation on the cognitive function of older people with subnormal cobalamin levels. Int J Geriatr Psychiatry 1998;13:611-616.
(66.) Teunisse S, Bollen AE, van Gool WA, Walstra GJ. Dementia and suboptimal levels of vitamin B12: effects of replacement therapy on dementia. J Neurol 1996;243:522-529.
(67.) Carmel R, Gott PS, Waters CH, et al. The frequently low cobalamin levels in dementia usually signify treatable metabolic, neurologic and electrophysiologic abnormalities. Eur J Heamatol 1995;54:245-253.
(68.) Levitt AJ, Karlinsky H. Folate, vitamin B12 and cognitive impairment in patients with Alzheimer's disease. Acta Psychiatr Scand 1992;86:301-305.
(69.) Miller JW, Green R, Mungas DM, et al. Homocysteine, vitamin B6, and vascular disease in AD patients. Neurology 2002;58:1471-1475.
(70.) Bottiglieri T, Godfrey P, Flynn T, et al. Cerebrospinal fluid S-adenosylmethionine in depression and dementia: effects of treatment with parenteral and oral S-adenosylmethionine. J Neurol Neurosurg Psychiatry 1990;53:1096-1098.
(71.) Morrison LD, Smith DD, Kish SJ. Brain S-adenosylmethionine levels are severely decreased in Alzheimer's disease. J Neurochem 1996;67:1328-1331.
(72.) Chambers JC, Ueland PM, Obeid OA, et al. Improved vascular endothelial function alter oral B vitamins. Circulation 2000;102:2479-2483.
(73.) Stamler J, Osborne J, Jaraki O, et al. Adverse effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. J Clin Invest 1993;91:308-318.
(74.) Christen Y. Oxidative stress and Alzheimer disease. Am J Clin Nutr 2000;71:621S-629S.
(75.) Ho PI, Ortiz D, Rogers E, Shea TB. Multiple aspects of homocysteine neurotoxicity: Glutamate excitotoxicity, kinase hyperactivation, and DNA damage. J Neurosci Res 2002;70:694-702.
(76.) Kruman II, Kumaravel TS, Lohani A, et al. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental model of Alzheimer's disease. J Neurosci 2002;22:1752-1762.
(77.) White AR, Huang X, Jobling MF, et al. Homocysteine potentiates copper- and amyloid beta peptide-mediated toxicity in primary neuronal cultures: possible risk factors in the Alzheimer's-type neurodegenerative pathways. J Neurochem 2001;76:1509-1520.
(78.) Ho PI, Collins SC, Dhitavat S, et al. Homocysteine potentiates amyloid beta neurotoxicity: role of oxidative stress. J Neurochem 2001;78:249-253.
(79.) Olney JW, Price MT, Salles KS, et al. L-homocysteic acid: an endogenous excitotoxic ligand of the NMDA receptor. Brain Res Bull 1987;19:597-602.
(80.) Parsons RB, Waring RH, Ramsden DB, Williams AC. In vitro effect of the cysteine metabolites homocysteic acid, homocysteine and cysteic acid upon human neuronal cell lines. Neurotoxicology 1998;19:599-604.
(81.) Kruman II, Culmsee C, Chan Al, et al. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J Neurosci 2000;20:6920-6926.
(82.) James SJ, Melnyk S, Pogribna M, et al. Elevation of S-adenosylhomocysteine and DNA hypomethylation: potential epigenetic mechanism for homocysteine-related pathology. J Nutr 2002; 132:2361 S-2366S.
(83.) West RL, Lee JM, Maroun LE. Hypomethylation of the amyloid precursor protein gene in the brain of an Alzheimer's disease patient. J Mol Neurosci 1995;6:141-146.
(84.) Rogaev EI, Lukiw WJ, Lavrushina O, et al. The upstream promoter of the beta-amyloid precursor protein gene (APP) shows differential patterns of methylation in human brain. Genomics 1994;22:340-347.
Alan Miller, ND--Technical Advisor, Thorne Research; Senior Editor, Alternative Medicine Review. Correspondence address: Thorne Research, PO Box 25, Dover, ID 83825 e-mail: alan@thorne.com
COPYRIGHT 2003 Thorne Research Inc.
COPYRIGHT 2003 Gale Group







 Home
Home A
A Macrodantin
Macrodantin